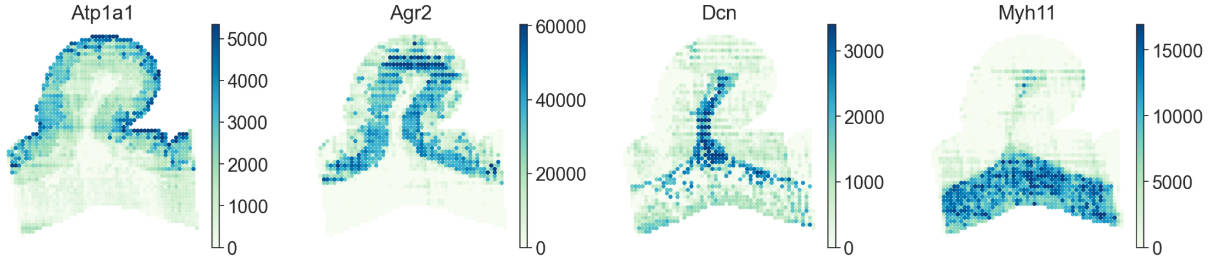
Reconstructing the spatial proteomics of rat large intestinal
To present the pipeline of Flow2Spatial more clearly, we show the rat large intestinal spatial proteomics reconstruction as an instance.
Here, we applied PLATO to an adult rat large intestinal villus-like tissue using a 25 µm chip. Nearly 2,000 proteins groups in ~100 channels (two angle) was successfully detected using LC-MS/MS, and needed to be reconstructed by Flow2Spatial. After quality control, the channel intensity used for reconstruction is located at https://github.com/bioinfo-biols/Flow2Spatial/tree/main/tests/df_pro_gut.csv.
Reconstruction
With the trained model weights (located at https://github.com/bioinfo-biols/Flow2Spatial/tree/main/tests), we can use F2S.model.reconstruction() to reconstruct spatial proteomics with the real MS values.
import Flow2Spatial as F2S
import pandas as pd
## Read protein abundance table
channel_intensity_list_dat = pd.read_csv('./df_pro_gut.csv')
channel_intensity_list_dat.loc[channel_intensity_list_dat['PG.Genes'].isna(), 'PG.Genes'] = channel_intensity_list_dat.loc[channel_intensity_list_dat['PG.Genes'].isna(), 'PG.ProteinAccessions'] + '_gene'
channel_intensity_list_dat_dropna = channel_intensity_list_dat.dropna()
## Prediction
Reconstruction = F2S.model.reconstruction(channel_intensity_list_dat_dropna, DNN_model='./Recontruct_weights_gut.pkl', Xchannels=57, mask='./mask')
Corresponding channel_intensity, DNN_model and mask locate at https://github.com/bioinfo-biols/Flow2Spatial/tree/main/tests. We can reconstruct protein spatial distribution with these files.
TroubleShooting: This step requires an internet connection for the first run. For non-networked servers, you can upload the required files (located in https://github.com/bioinfo-biols/Flow2Spatial/tree/main/Flow2Spatial/model/resnet34-b627a593.pth) to the model folder under the Flow2Spatial installation path.
In default, an AnnData object would be saved at “./save_environ/adata.h5ad” (located at https://github.com/bioinfo-biols/Flow2Spatial/tree/main/tests/adata.h5ad). You are welcome to use h5ad readers, such as sc.read_h5ad('save_environ/adata.h5ad') in scanpy, for further spatial proteomics analysis.
## For example
import scanpy as sc
import seaborn as sns
sns.set(style="ticks", font_scale=2)
adata = sc.read_h5ad('./save_environ/adata.h5ad')
sc.pl.spatial(adata, color=['Atp1a1', 'Pdia3', 'Dcn', 'Myh11'], spot_size=1.2, cmap='GnBu', vmax='p99', wspace=0., frameon=False, show=False)